KRAS and Its Importance
Indications
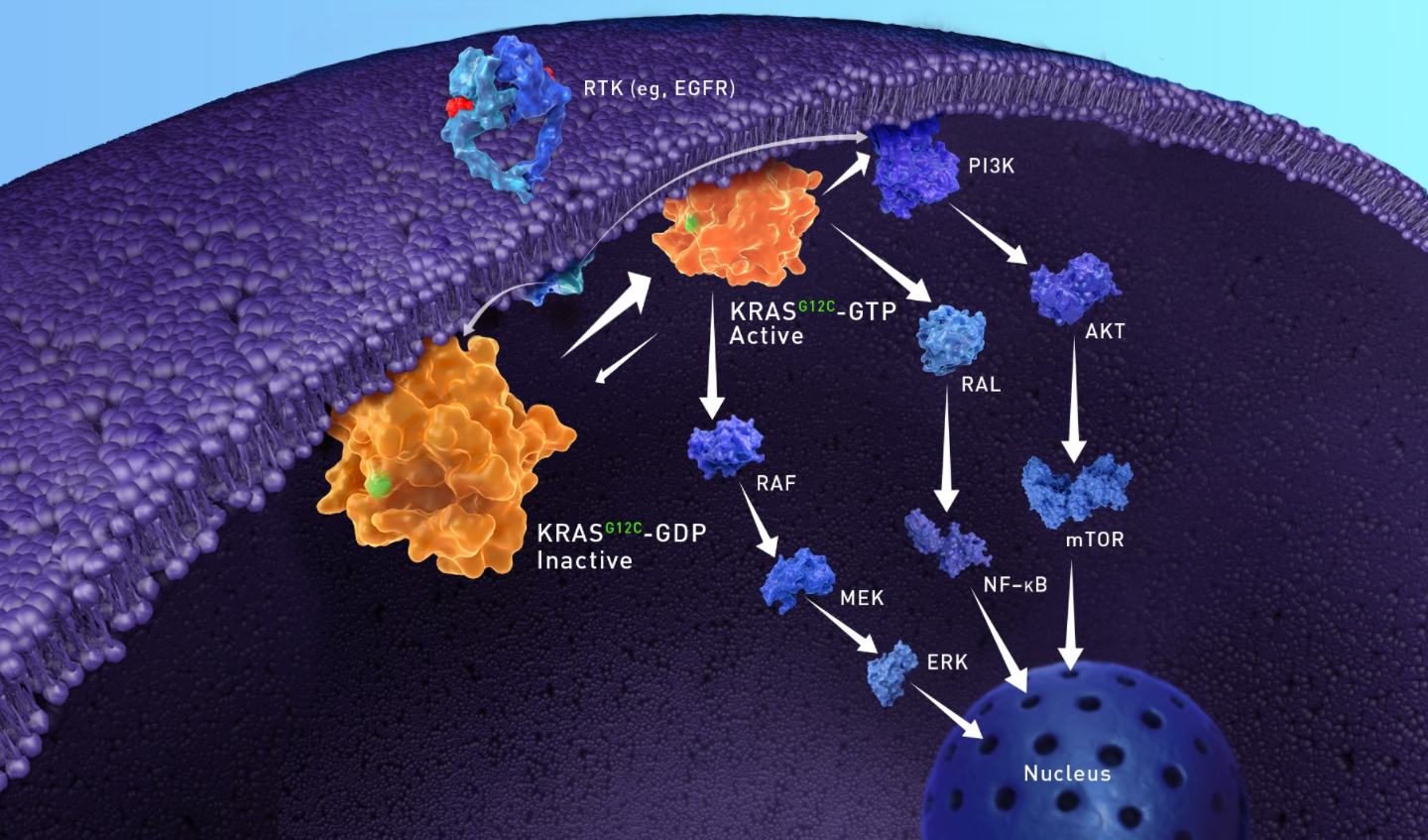
The inhibition of KRAS G12C dependent cancers primarily addresses several types of cancers where this specific mutation is prevalent. These include:
- Non-Small Cell Lung Cancer (NSCLC): A common type of lung cancer where KRAS G12C mutations are frequently found.
- Pancreatic Ductal Adenocarcinoma (PDAC): A prevalent mutation in this aggressive form of pancreatic cancer.
- Colorectal Cancer: KRAS G12C mutations are also present in a subset of these cases.
This targeted therapeutic strategy employs covalent inhibition, exemplified by adagrasib (Krazati), which specifically addresses the G12C mutation. This medication signifies a substantial progression in oncology, providing novel therapeutic alternatives for patients with these mutations.
Biological Role of KRAS
Primary Biological Roles
- Cell Growth and Proliferation: KRAS helps control when cells divide and multiply, which is essential for tissue development, repair, and maintenance.
- Cell Differentiation: It influences how cells specialize into different types (e.g., during embryonic development or wound healing).
- Cell Survival and Apoptosis: KRAS signaling can prevent programmed cell death (apoptosis), ensuring cells survive under normal conditions.
- Cytoskeletal Regulation and Migration: It plays a role in cell movement and structure, which is important for processes like immune responses or embryonic development.
Why Target Was Chosen
- KRAS was chosen as a cancer target due to its role as a key oncogene that is mutated in 20–25% of all cancers, driving uncontrolled cell growth via persistent signaling pathways.
- It’s especially prevalent in deadly types like pancreatic (up to 90%), colorectal (40-50%), and non-small cell lung cancer (30%), where it acts as a driver mutation causing treatment resistance.
- Long deemed “undruggable” for lacking binding sites, breakthroughs enabled inhibitors like sotorasib and adagrasib to treat G12C mutations, offering tumor shrinkage in 30–40% of patients.
- Targeting KRAS addresses unmet needs in high-mortality cancers, with ongoing research expanding to other mutations and combinations for broader impact.
Intended Audience or Stakeholder for This Target
(e.g., internal leadership, partners, or external funding bodies)
Charles M. Baum, M.D., Ph.D. (Founder, President, and CEO): A pivotal figure, Baum joined Mirati in 2012 and became CEO in 2013. He led the company’s focus on targeted oncology therapies, including the KRAS program. Under his leadership, Mirati raised significant funding and advanced multiple drug candidates.
Isan Chen, M.D. (Executive Vice President and Chief Medical Officer): Chen was key in clinical development, overseeing trials for drugs like adagrasib. His expertise in oncology helped shape Mirati’s drug discovery strategies.
Jamie Christensen, Ph.D. (Executive Vice President and Chief Scientific Officer): Christensen drove the scientific aspects of drug discovery, including preclinical research on KRAS inhibitors. He was involved in Mirati’s campaigns from around 2017 onward.
Board of Directors: Included influential members like
- Faheem Hasnain (Chairman, experienced biotech executive from Receptos and Facet Biotech).
- Other notable directors: Julie Cherrington (biotech veteran), Bruce Carter (former Novo Nordisk exec), and Maya Martinez-Davis (from Merck KGaA).
- The board provided oversight on R&D investments and partnerships.
Krazati™ Hit-Finding Phase
Chemical Starting Point
The initial central scaffold was arrived at from a patent-busting exercise of the patent entitled “INHIBITORS OF KRAS G12C,” patent application: WO 2015/054572 A1, Applicant: ARAXES PHARMA LLC
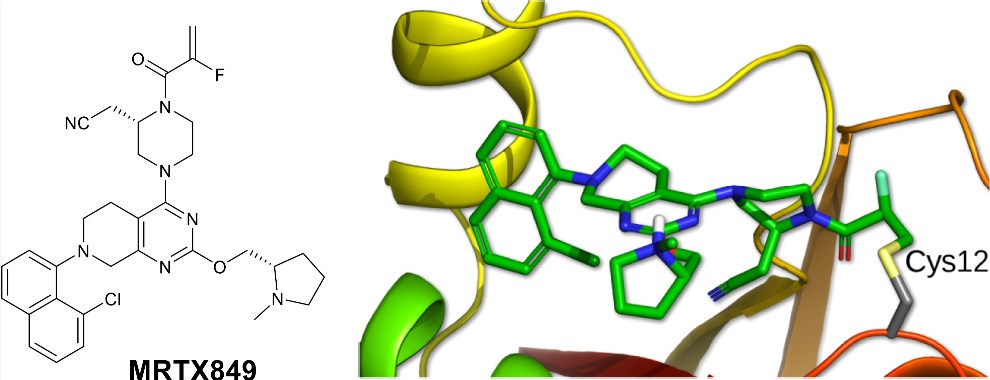
Lead Identification
Novel binding was identified via in silico drug design employing published X-ray co-crystal structures from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB), accessible as .pdb files: https://www.rcsb.org/. New compounds targeting KRAS G12C, developed outside existing patent estates, were informed by proprietary in-house x-ray co-crystal structures provided by collaborator Array Biopharma to direct subsequent designs.
Key Considerations or Constraints Guided by Early Development Work
Early development was focused on developing chemical matter not covered by existing composition of matter patents. Activity against the known KRAS G12C mutant immortalized MIA PaCa-2 and NCI-H358 cell lines, as these cell lines harbored the KRAS G12C mutation, and time-dependent modification of the KRAS G12C recombinant protein were key considerations. Finally, the development of robust synthetic routes to enable structure-based in silico designs was another key consideration.
How Compound Was Advanced from Hit to Lead Drug Candidate
- What structure-activity relationship (SAR) trends helped guide optimization?
- How did you address challenges like potency, solubility, or metabolic stability?
- Were there any innovative approaches or breakthrough moments during this phase?
The key structure-activity relationships (SAR) include:
- Hydroxyl Moiety Removal: Removal of the hydroxyl group from compound 1 improved oral bioavailability and reduced clearance by minimizing conjugative metabolism (e.g., glucuronidation and sulfation).
- Naphthol Replacement: Substituted indazoles were explored as replacements for the hydroxy naphthyl group to maintain hydrophobic pocket binding and hydrogen bonding with Asp69.However, these replacements showed poor cellular permeability and were potent P-glycoprotein efflux substrates.
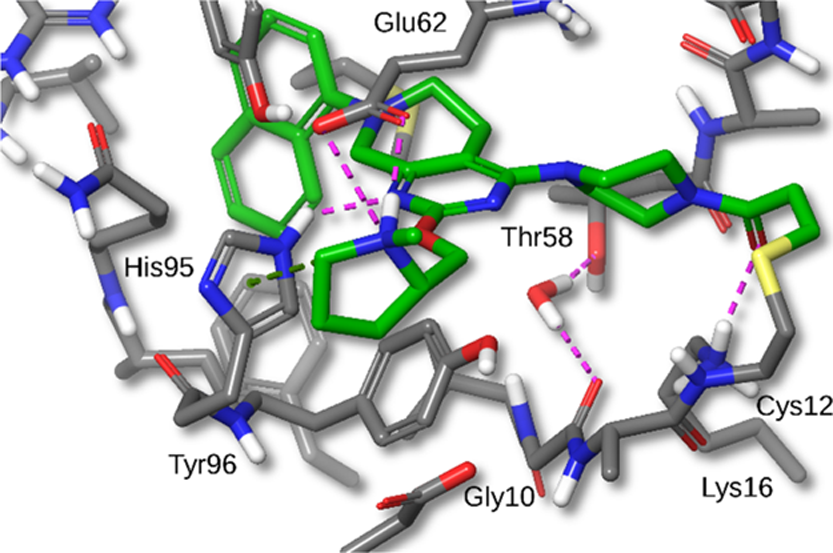
- Bound Water Displacement: The deshydroxy analogue 7 revealed a bound water molecule near Gly10 and Thr58.Displacement of this water with a cyanomethyl-substituted piperazine (compound 12a) significantly increased potency by forming new hydrogen bonding interactions with KRASG12C.
- 8-Position Substitution on Naphthyl Ring: Substitutions at the 8-position of the naphthyl ring (e.g., chloro and methyl groups in compounds 18 and 19) improved potency, with cellular IC50 values of 1 nM.These substitutions filled a small hydrophobic pocket formed by residues Val9, Thr58, Met72, and Tyr96.
- Electrophile Substitution: Substituting the acrylamide warhead with a 2-fluoroacrylamide (compound 20, MRTX849) reduced glutathione (GSH) conjugation and improved whole blood stability (>50 h across species) while maintaining cellular potency (IC50 = 5–14 nM).
These SAR insights guided the optimization of MRTX849 as a potent, selective, and orally bioavailable KRASG12C inhibitor.
Learned Key Points from Pharmacokinetics and Pharmacodynamics (PK/PD)
What Body of Drug Did
Pharmacokinetic (PK) Data
- MRTX849 Across Species:
CLEARANCEHALF-LIFEVOLUMEMouse (3 mg/kg IV):Clearance (CL): 19.9 mL/min/kgHalf-life (t1/2): 1.51 hVolume of distribution (VSS): 2.02 L/kg
Rat (3 mg/kg IV):CL: 44.2 mL/min/kgt1/2: 2.57 hVSS: 20.7 L/kg
Dog (3 mg/kg IV): CL: 29.6 mL/min/kgt1/2: 7.56 hVSS: 17.4 L/kg
Oral Bioavailability (30 mg/kg PO):Mouse: 62.9%Rat: 29.7%Dog: 25.9%
Metabolism:MRTX849 showed reduced glutathione (GSH) conjugation compared to earlier analogues. Metabolite profiles were similar across species, with oxidation primarily occurring on the methylpyrrolidine group.
| Species | CL (mL/min/kg) | t½ (h) | Vss (L/kg) | F (%) |
| Mouse | 19.9 | 1.51 | 2.02 | 62.9% |
| Rat | 44.2 | 2.57 | 20.7 | 29.7% |
| Dog | 29.6 | 7.56 | 17.4 | 25.9% |
What the Drug Did to the Body
Pharmacodynamic (PD) Data
KRASG12C Target Engagement:
- MRTX849 demonstrated dose-dependent covalent modification of KRASG12C in NCI-H358 xenograft-bearing mice.
- Maximal target engagement was achieved at a 100 mg/kg dose.
Results in Metabolism or Exposure
Results in Animal Models
Efficacy and Safety Data Observed
All mice in the 100 mg/kg cohort remained tumor-free for the duration of the study, and 2 of 7 mice in the 30 mg/kg cohort remained tumor-free through study day 70. In addition, drug treatment was well-tolerated with no loss of body weight at any dose level.
Drug Behavior in Vivo
Anti-Tumor Efficacy
MIA PaCa-2 Xenograft Model:
- 10 mg/kg dose: Tumor regressions were observed, but rapid rebound occurred after dosing ended.
- 30 mg/kg dose: Significant tumor regression with durable responses in 2 of 7 mice through study day 70.
- 100 mg/kg dose: Complete tumor regression with all mice remaining tumor-free for the 70-day monitoring period.
Did this data support moving into IND-enabling studies or clinical development?
YES!